
Generalization of CHARMM Molecular Force-Field to Small Molecules
Published:
A typical biomolecule like protein, lipid membrane, cholesterol has typically more than 1000 atoms. Including local environments like water and ions adds typically more than 10000 atoms to the system. Handling such large system requires very simple potential function. Additionally, we are often interested in studying interaction of small molecules with biomolecules. Atomistic force-field defines molecular potential in terms of empirical functions for bonded and non-bonded components. To ensure correct behavior of the empirical functions they are generally fitted against quantum mechanically derived target data such as dipole moment, molecular polarizability, vibrational spectra, potential energy scans. But ultimate goal of any force- field is to correctly represent bulk properties. that My research interest includes methodological advances of chemical phenomena. I want to use my expertise of computing, quantum chemistry and molecular mechanics to improve the accuracy and precision of current drug and catalyst design protocols. This involves formulation, development, implementation and application of QM mediated free energy estimation methodologies as well as ease of use of the entire computational exercise. The scope of this work involves use of a range of chemical models with increasing complexity such as molecular docking, molecular mechanics and quantum mechanics. I have worked on each of these components during my research career as PhD and Postdoc. Following is brief description of key areas which I have been worked in.
Optimization of CHARMM Additive and Drude Polarizable Force-field
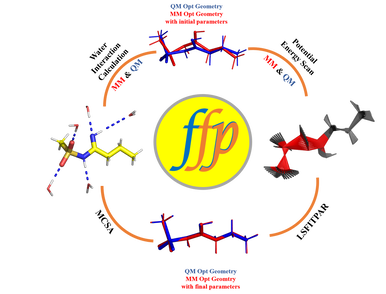 CHARMM force-field describes the potential energy of a molecule in terms of bond, angle, dihedral, improper, electrostatic and van der Waals parameters. This force-field can be broadly categorized into Additive and Drude FF, the latter of which includes polarizabilities along with atomic charges as a part of electrostatic parameters. It contains carefully optimized parameters for biological systems, including nucleic acids, proteins, lipids etc., such that both microscopic and macroscopic properties are well-reproduced. However, parametrization of a new molecular system from first principles is a tedious and exhaustive process which usually requires lot of scripts and programs. To simplify the process, we developed a standalone python package FFParam which provides one platform to assist various aspects of parameterization process. It can handle both CHARMM Additive and Drude parameterization process wherein it can create, run, extract and analyze QM data from Gaussian and Psi4 and MM data from CHARMM and OpenMM. FFParam has cross-platform compatible GUI which assists display of text, plot, and 3D- molecular geometry.
CHARMM force-field describes the potential energy of a molecule in terms of bond, angle, dihedral, improper, electrostatic and van der Waals parameters. This force-field can be broadly categorized into Additive and Drude FF, the latter of which includes polarizabilities along with atomic charges as a part of electrostatic parameters. It contains carefully optimized parameters for biological systems, including nucleic acids, proteins, lipids etc., such that both microscopic and macroscopic properties are well-reproduced. However, parametrization of a new molecular system from first principles is a tedious and exhaustive process which usually requires lot of scripts and programs. To simplify the process, we developed a standalone python package FFParam which provides one platform to assist various aspects of parameterization process. It can handle both CHARMM Additive and Drude parameterization process wherein it can create, run, extract and analyze QM data from Gaussian and Psi4 and MM data from CHARMM and OpenMM. FFParam has cross-platform compatible GUI which assists display of text, plot, and 3D- molecular geometry.
Development of Machine learning model to train charges and polarizabilities
 CHARMM Drude force-field uses charged auxiliary (Drude) particles attached to the non-hydrogen atoms via a harmonic spring to address electronic degree of freedom. Development of automated tool to predict electrostatic parameters of Drude polarizable FFs is quite challenging, as it requires a balance of partial atomic charges, atomic polarizabilities, Thole scale factors, and treatment of lone pairs. We developed a machine learning model to estimate QM-level RESP atomic charges, atomic polarizability and Thole scale factors (which has no QM analog). The approach includes the calculation of atomic polarizabilities and partial atomic charges of about 50K molecules (including 900 FDA approved drugs) using QM-based methods to produce a large training-set, which is be used to build and optimize the machine learning model for rapid parameter estimation.
CHARMM Drude force-field uses charged auxiliary (Drude) particles attached to the non-hydrogen atoms via a harmonic spring to address electronic degree of freedom. Development of automated tool to predict electrostatic parameters of Drude polarizable FFs is quite challenging, as it requires a balance of partial atomic charges, atomic polarizabilities, Thole scale factors, and treatment of lone pairs. We developed a machine learning model to estimate QM-level RESP atomic charges, atomic polarizability and Thole scale factors (which has no QM analog). The approach includes the calculation of atomic polarizabilities and partial atomic charges of about 50K molecules (including 900 FDA approved drugs) using QM-based methods to produce a large training-set, which is be used to build and optimize the machine learning model for rapid parameter estimation.